


























کشف روش کوانتومی برای تسریع طراحی داروهای نوری

برنا - گروه علمی و فناوری: محققان با استفاده از ترکیب هوش مصنوعی و محاسبات کوانتومی، روش جدیدی برای کشف سریعتر مواد فوتوکرومیک معرفی کردند که میتواند انقلاب بزرگی در داروشناسی نوری ایجاد کند.
یک روش جدید ترکیب کوانتومی-کلاسیکی سرعت کشف مواد فوتوکرومیک را افزایش میدهد.
به گزارش ساینس دیلی، یک تیم تحقیقاتی مشترک یک رویکرد نوآورانه کوانتومی-کلاسیکی برای طراحی مواد فوتوکرومیک (ترکیبات حساس به نور) توسعه دادهاند که ابزاری قدرتمند برای تسریع کشف مواد فراهم میآورد. یافتههای این تحقیق در مجله Intelligent Computing منتشر شده است.
در ادامه تحقیقات پیشین خود در همین مجله، محققان روش پایهگذاری محاسباتی-کوانتومی و کاهش متغیرهای کوانتومی را به عنوان مبنای این رویکرد معرفی کردند.
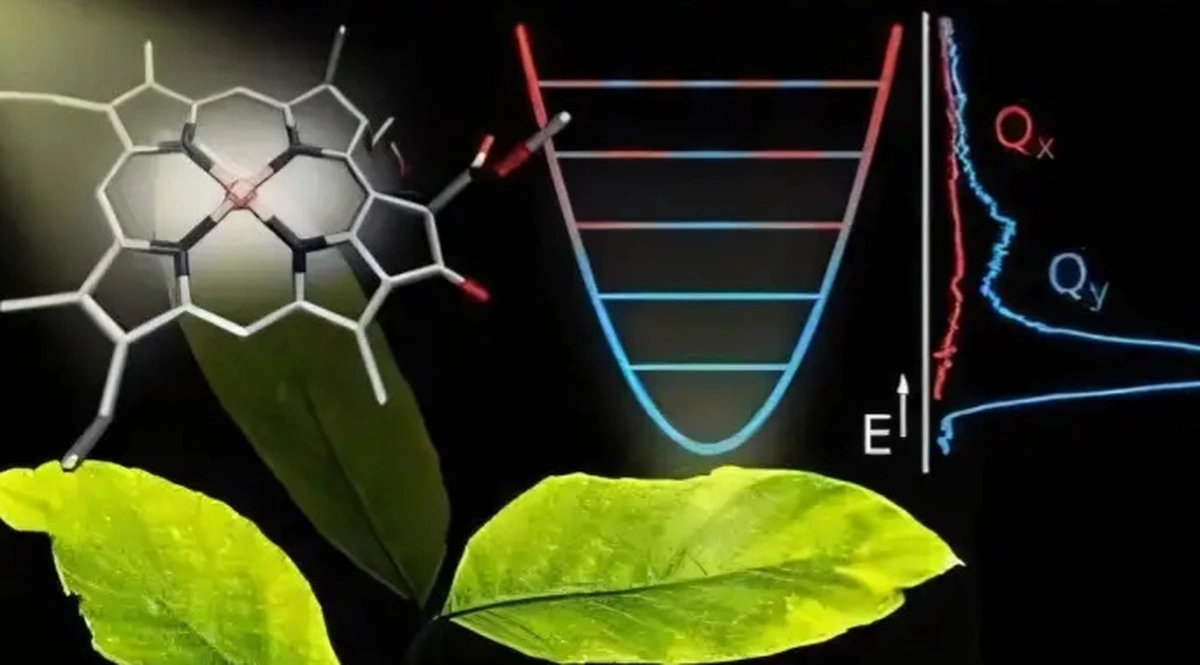
برای ارزیابی اثربخشی این روش، تیم تحقیقاتی یک مطالعه موردی در زمینه داروشناسی نوری (Photopharmacology) انجام دادند و ۴۰۹۶ مشتق دیاریلاتن را بررسی کردند. آنها پنج کاندیدای امیدوارکننده را شناسایی کردند که دو ویژگی حیاتی را نشان دادند: حداکثر طول موج جذب بزرگ و قدرت نوسانی بالا. این ویژگیها برای کاربردهایی همچون تحویل دارو تحت کنترل نور در داروشناسی نوری حائز اهمیت هستند.
داروشناسی نوری: زمینهای نوین در پزشکی
داروشناسی نوری یک شاخه نوظهور در علم پزشکی است که از نور برای فعال یا غیرفعال کردن مولکولهای خاص استفاده میکند و امکان تحویل هدفمند دارو را فراهم میآورد. در میان مواد استفادهشده برای این منظور، مشتقات دیاریلاتن به دلیل تغییر رنگ خود در پاسخ به نور و پایداری در دماهای مختلف، بهویژه امیدوارکننده هستند.
کشف مواد با هدایت هوش مصنوعی و محاسبات کوانتومی
برای شناسایی مشتقات بهینه دیاریلاتن، تیم تحقیقاتی ابتدا ساختارهای مولکولی را تولید کرده و محاسبات شیمی کوانتومی را روی ۳۸۴ مشتق دیاریلاتن انجام دادند تا ویژگیهای آنها را پیشبینی کنند. سپس از نتایج این محاسبات برای آموزش یک مدل یادگیری ماشینی استفاده کردند تا ویژگیهای مجموعهای از ۴۰۹۶ مشتق را پیشبینی کنند.
در ادامه، از یک کامپیوتر کوانتومی برای بهینهسازی این پیشبینیها استفاده شد تا مولکولهایی با بزرگترین طولموجهای جذب حداکثر شناسایی شوند، با استفاده از مدل ایزینگ همیلتونی، یک مدل ریاضی که برای توصیف سیستمها به کار میرود. در نهایت، محاسبات شیمی کوانتومی روی کامپیوترهای کلاسیکی انجام شد تا ویژگیهای کاندیداهای برتر بررسی و تأیید شوند.
بهینهسازی و تأیید کوانتومی
در فرایند بهینهسازی کوانتومی، از محاسبات ۱۲ کیوبیتی کوانتومی برای شبیهسازی مؤثر حالت پایه (کمترین حالت انرژی) و چهار حالت برانگیخته از مدل ایزینگ استفاده شد. این مرحله به شناسایی مشتقات دیاریلاتن با بزرگترین و دومین تا چهارمین طول موجهای جذب کمک کرد. سپس، محاسبات شیمی کوانتومی برای تحلیل نقش اوربیتالهای مولکولی در جذب استفاده شد. این مرحله راهنمای طراحی مشتقات جدید دیاریلاتن است که نه تنها طولموجهای جذب بزرگی دارند، بلکه قدرت نوسانی بالایی نیز دارند.
زمانی که این روش روی شبیهساز کوانتومی آزمایش شد، نتایج آن با نتایج بهدستآمده از eigsolver دقیق، ابزاری محاسباتی برای محاسبه سطوح انرژی سیستم همیلتونی (در این مورد مدل ایزینگ)، همخوانی خوبی نشان داد. جالب اینجاست که حتی در دستگاههای واقعی کوانتومی، این روش نتایج دقیقی مشابه به شبیهساز نشان داد، که به دلیل تکنیکهای پیشرفته سرکوب و کاهش خطا بوده است.
در سالهای اخیر، ترکیب محاسبات شیمی کوانتومی با یادگیری ماشینی پتانسیل زیادی در تسریع کشف مواد نوین نشان داده است. در حالی که این روش ترکیبی منابع و زمان کمتری نسبت به روشهای سنتی مصرف میکند، هنوز چالشهای اساسی پیشرو وجود دارد، از جمله محدودیتها در اندازه و کیفیت دادههای آموزشی و دشواریها در کاوش مؤثر فضاهای شیمیایی بزرگ با استفاده از تکنیکهای بهینهسازی گسسته. روش کوانتومی-کلاسیکی جدید ثابت کرده است که قادر است این چالشها را برطرف کند و امکان کشف انواع دیگر مواد مفید را در آینده فراهم کند.
انتهای پیام/


برنا گزارش میدهد؛









برنا گزارش میدهد؛







نظر شما

پیشنهاد سردبیر












پرونده ویژه







